
Translate this page into:
West Syndrome due to ALG13 mutation presenting vigabatrin-associated reversible MRI signal changes and drug-induced dyskinesia
 , Brenda Souza de Oliveira Reis2, Gustavo Leite Franklin3,, Bernardo Corrêa de Almeida Teixeira4, Ana Chrystina de Souza Crippa5
, Brenda Souza de Oliveira Reis2, Gustavo Leite Franklin3,, Bernardo Corrêa de Almeida Teixeira4, Ana Chrystina de Souza Crippa5

*Corresponding author: Gustavo Leite Franklin, Department of Internal Medicine, Pontifical Catholic University of Paraná, Curitiba, Brazil. gustavolf_88@hotmail.com
-
Received: ,
Accepted: ,
How to cite this article: Souza LP, Reis BS, Franklin GL, Teixeira BC, Crippa AC. West Syndrome due to ALG13 mutation presenting vigabatrin-associated reversible MRI signal changes and drug-induced dyskinesia. Med India. 2024;3:33-6. doi: 10.25259/MEDINDIA_27_2023
Abstract
Heterozygous mutations in the asparagine-linked glycosylation 13 (ALG13) gene cause a rare genetic disorder that severely affects neurological development. Symptoms associated with ALG13 mutations include severe epilepsy and/or West syndrome (WS). Most cases reported in the literature describe typical characteristics of patients within whom de novo mutations have been identified. A 5-month-old girl with developmental delay and epileptic spasms, without dysmorphic features, is described. Electroencephalography (EEG) showed hypsarrhythmia, compatible with WS, but complementary examinations did not find an underlying etiology. Vigabatrin therapy was started, and then adrenocorticotropic hormone (ACTH) therapy was added. On follow-up, brain magnetic resonance imaging (MRI) revealed restricted diffusions compatible with vigabatrin-associated reversible MRI signal changes. As the spasms and hypsarrhythmia on EEG persisted, a second cycle of ACTH was performed, and then, the patient manifested a hyperkinetic movement, characteristic of drug-induced dyskinesia. Further investigation was carried out, and whole-exome sequencing revealed a de novo pathogenic variant in ALG13 gene. The current case report expands our knowledge of WS by considering a female patient with unspecific clinical features arising from an undefined subjacent etiology. This case also emphasizes the importance of a thorough work-up to determine the underlying genetic etiology and presents the challenges in the diagnostic investigation in clinical practice.
Keywords
West syndrome
Infantile spasms
X-linked infantile spasms
Epileptic seizures

INTRODUCTION
Asparagine-linked glycosylation 13 (ALG13) deficiency is a rare X-linked inborn metabolic disorder displaying severe neurological delay.[1] ALG13 may be related to a congenital disorder of glycosylation, featuring cranial dysmorphism, microcephaly, intellectual disability, and seizures.[1,2] At present, most reports of ALG13 deficiency involve females with the ALG13 NM_001099922:exon3:c.A320G: p.N107S.[1] A heterozygous de novo mutation produces a range of clinical features that frequently involve developmental delay and epileptic conditions in girls.[1,2] Therefore, understanding the clinical presentations associated with the treatment response can improve our knowledge and the diagnosis of West syndrome (WS) forms. Here, it describes a challenging diagnosis and treatment pathway of a girl with WS due to ALG13 mutation, which presented vigabatrin-associated reversible brain magnetic resonance imaging (MRI) signal changes and also developed drug-induced hyperkinetic movements during the treatment optimization.
CASE REPORT
A 5-month-old girl presented with tonic spasms at 4 months of age, occurring in clusters of 20–40/day, associated with developmental regression. On clinical examination, the child demonstrated global hypotonia, but did not present facial dysmorphisms nor other clinical-specific features. The child was born by natural delivery (Apgar, 9/9), the parents were not consanguineous, and there was no familial history of epilepsy. An initial laboratory screening was unremarkable. Electroencephalogram (EEG) found a hypsarrhythmia pattern, compatible with the diagnosis of WS. The first brain MRI at 5-month-old revealed no abnormalities. At that time, it was started the initial treatment with vigabatrin (50 mg/kg/day), which led to no response. Thereafter, the vigabatrin dose was adjusted to 150 mg/kg/day. However, the patient failed to respond to vigabatrin optimization. Thus, synthetic adrenocorticotropic hormone (ACTH) therapy, at 0.5 mg/kg/day was added to the therapy (corresponding to 20 IU/kg/day of natural ACTH). On the 3rd day of the therapy, the spasms and hypsarrhythmia subsided, and developmental gains were subsequently observed. However, two months after initiating ACTH therapy, there was a significant return of tonic spasms. In this way, the patient underwent further clinical evaluation, including additional MRI, which revealed restricted diffusion in the anteroinferior portions of the hypothalamus and posterior tegmental tracts of the pons, and hyperintense lesions in the anteroinferior portions of the hypothalamus and posterior tegmental tracts of the pons on T2-weighted image [Figure 1], compatible with vigabatrin-associated MRI signal changes. To control the tonic spasms, a second cycle of ACTH therapy, in the same dose, was performed. After this cycle of treatment, hyperkinetic movements were identified, in which the patient presented involuntary and repetitive mouth opening, facial grimacing, and asymmetric contractions of the limbs and tongue. There was no loss of consciousness or epileptiform discharges on EEG during these events, compatible with drug-induced dyskinesia.[3] Although it was not clear if the vigabatrin or the association of ACTH with vigabatrin was the reason for the induced movements, it led to the necessity to interrupt vigabatrin. Therefore, a genetic investigation was carried out, and whole-exome sequencing prompted the diagnosis of a de novo pathogenic variant NM_001099922:exon3:c.A320G: p. N107S in the ALG13 gene, compatible with developmental epileptic encephalopathy 36 (DEE36; OMIM#300884).
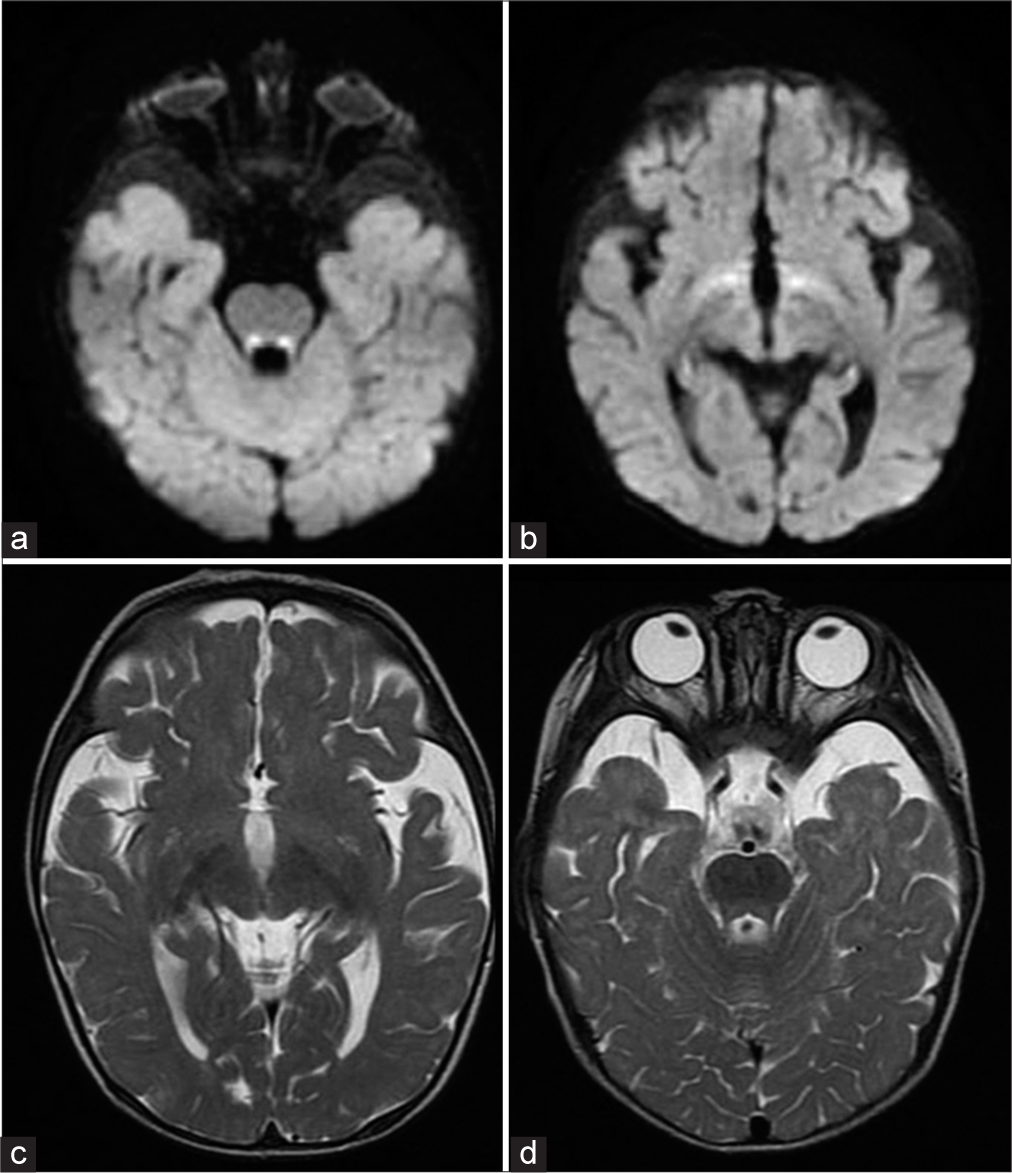
- (a and b) Brain magnetic resonance imaging (MRI) showing restricted diffusion in the anteroinferior portions of the hypothalamus and posterior tegmental tracts of the pons. (c and d) T2-weighted brain MRI showing hyperintense lesions in the anteroinferior portions of the hypothalamus and posterior tegmental tracts of the pons.
DISCUSSION
A 5-month-old female with severe epileptiform encephalopathy and developmental delay, diagnosed with WS was described in this case report. The infantile spasm onset was consistent with the description of other patients with WS, aged 4–6 months.[4] Symptomatic WS forms are most common, occurring in combination with an underlying disease.[5] Initially, a specific etiology was not considered in this case, because the patient had no history of gestational or delivery problems, coagulation disturbances, neuroimage abnormalities, or dysmorphic features differing from those of prior published reports.[1,4] Thus, additional evaluation was performed after the failure of the first therapies, finding the underlying gene. A rigorous evaluation with neuroimaging and genetic testing may provide an etiological diagnosis in most patients with WS. Chourasia et al. described a cohort with 127 patients, in which it was found that when an identifiable genetic diagnosis was present, the absence of a recognizable dysmorphic/syndromic diagnosis was associated with a poorer treatment outcome.[6]
Most cases of ALG13 deficiency occur among young females displaying motor development delays and epileptic spasms;[1] similar features are observed in those with DEE. Although features including dysmorphic facial features (42%), ocular defects (46%), bone abnormalities (42%), and movement disorders (41%) occur inconsistently in patients with ALG13 deficiency,[7] they are highly suggestive of genetic disease. Furthermore, a prominent jaw, smooth philtrum, bulbous nose, large mouth and ears, thin upper lid, widely spaced teeth, and high palate have been described in young females with c.320A>ALG13 mutations.[1]
Reversible MRI findings during treatment of infantile spasms with vigabatrin have been well documented.[8-10] The pathophysiology and the anatomical correlation are not so certain according to the literature, but descriptions, such as this one, may help to better understand this situation in the future. These findings are largely asymptomatic, although some patients may present with hyperkinetic movement disorders.[10] These movements are mainly choreiform or dystonic movements without an EEG correlation. Risk is associated with a non-cumulative, but high peak dose. It is not clear if ACTH therapy may enhance the risk of drug-induced dyskinesia.[3] In our patient, dyskinesia developed during concomitant therapy with vigabatrin (150 mg/kg/day) and ACTH (0.5 mg/kg/day). Although dyskinesias were not evident until the addition of ACTH, these movements are subtle and may be easily confounded with epileptic seizures. Our analysis is that the combination of those drugs may be a greater predisposing factor than the use of vigabatrin isolated. This vision occurred in other cases in which there was the addition of ACTH to vigabatrin.[3] This discussion is important, and quick identification of these movement disorders is essential since the treatments would be carried out in totally opposite directions. Most children with ALG13 deficiency seem to respond satisfactorily to primary therapy. Hormonal drugs (ACTH or prednisone) are described as more effective than vigabatrin for treating WS in infants with ALG13 deficiency.[5,7] In our case, the patient responded only after the second ACTH cycle, which is unusual in ALG13 reports. Important to mention that, in WS, responsiveness to therapy is influenced by several variables, including ACTH dosage, disease etiology, and likely broad phenotypes of genetic syndromes. Nonetheless, a diagnosis should be made as soon as possible to avoid detrimental developmental delays. In the long term, most children with WS with genetic etiologies develop severe epilepsy and neurological delays.[7]
CONCLUSION
A 5-year-old girl with WS was described, in which no underlying cause was identified initially. The patient was resistant to the first treatments and adjustments, and the findings of a vigabatrin-associated reversible MRI signal change and the development of drug-induced dyskinesia led to a challenging diagnosis pathway. A pathogenic variant in ALG13 gene was found, and the patient responded after a second ACTH cycle. This case emphasizes the importance of a thorough work-up to determine the underlying genetic etiology in WS.
Ethical approval
Ethical Committee of Research of Universidade Federal do Paraná (a61921316.4.3002.0096 and 61921316.4.0000.0103 CAAE), dated April 6, 2023.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- ALG13 X-linked intellectual disability: New variants, glycosylation analysis, and expanded phenotypes. J Inherit Metab Dis. 2021;44:1001-12.
- [CrossRef] [PubMed] [Google Scholar]
- Epileptic encephalopathies: Clinical aspects, molecular features and pathogenesis, therapeutic targets and translational opportunities, and future research directions. J Child Neurol. 2018;33:7-40.
- [CrossRef] [PubMed] [Google Scholar]
- Adrenocorticotropic hormone (ACTH)-induced dyskinesias in infantile spasms: A video case report. Am J Case Rep. 2022;23:e935349.
- [CrossRef] [PubMed] [Google Scholar]
- West syndrome: A review and guide for pediatricians. Clin Drug Investig. 2018;38:113-24.
- [CrossRef] [PubMed] [Google Scholar]
- Safety and effectiveness of hormonal treatment versus hormonal treatment with vigabatrin for infantile spasms (ICISS): A randomized, multicentre, open-label trial. Lancet Neurol. 2017;16:33-42.
- [CrossRef] [PubMed] [Google Scholar]
- Infantile spasms: Assessing the diagnostic yield of an institutional guideline and the impact of etiology on long-term treatment response. Epilepsia. 2022;63:1164-76.
- [CrossRef] [PubMed] [Google Scholar]
- Predominant and novel de novo variants in 29 individuals with ALG13 deficiency: Clinical description, biomarker status, biochemical analysis, and treatment suggestions. J Inherit Metab Dis. 2020;43:1333-48.
- [CrossRef] [PubMed] [Google Scholar]
- Treatment of West syndrome with vigabatrin: Reversible MRI signal changes. Arq Neuropsiquiatr. 2011;69:998.
- [CrossRef] [PubMed] [Google Scholar]
- Vigabatrin-associated reversible MRI signal changes in patients with infantile spasms. Epilepsia. 2010;51:1297-304.
- [CrossRef] [PubMed] [Google Scholar]
- Risk of vigabatrin-associated brain abnormalities on MRI in the treatment of infantile spasms is dose-dependent. Epilepsia. 2017;58:674-82.
- [CrossRef] [PubMed] [Google Scholar]




